最初のヒトゲノム解読以来、私たちが学んだ五つのこと
Blog Post
カテゴリー:遺伝
By Mark Wanner

1. 完全は必ずしも完全を意味するわけではない
最初の「完全な」ヒトゲノム配列、およびその後の大多数のゲノム配列では、実際には完全な配列の10%近くが省略されています。染色体には、セントロメア (染色体が凝縮する「x」字型の中央) や各染色体の端にあるテロメアなど、標準的な方法を使用して配列を決定するのが非常に困難な部分があります。ほかにも、参照配列に対して正確に位置合わせすることが不可能な、反復性の高い領域が存在し、これらの領域は省略されています。およそ20年前、最初の配列が完成したと報告された時、タンパク質をコードしていないゲノムの98.5%について、その重要性と機能の可能性に関して多くの議論が行われており、特定の非コード領域を研究対象から除外することは合理的だと思われました。現在では、これらの配列が実際に非常に重要である可能性があることがわかっており、T2Tコンソーシアムなどの最近の取り組みでは、改良されたロングリード配列決定技術(下記 No. 4を参照)を使用して、ついにそのギャップを埋めることができました。
2. 一つのゲノムは、私たちが思っているほど多くのことを私たちに教えてくれない
最初のヒトゲノム配列は、米国ニューヨーク州バッファロー出身の北欧系住民の男性から提供された検体から生成されました。これは「生命の青写真」として賞賛され、当時は、さらなる配列が完成して分析される前であっても、健康と病気に関する重要な知見を提供するには十分であると考えられていました。しかし、実際はそうではありませんでした。今にして思えば、その理由は明らかなのですが、単一のデータセットでは、地球上の他のすべてのヒト集団と同様に、男女のうち一つの性が完全に除外されます。性別や民族によって病気の感受性やリスクが異なることは長い間知られていましたが、ゲノムデータセットは多様化する必要があるというメッセージの認知を高めるには時間がかかりました。そして、状況は改善したとはいえ、配列決定されたゲノムのほとんどは依然として北欧の祖先のものです。つまり、やるべきことがまだたくさん残っているのです。
3. 機能の喪失が必ずしも悪影響をもたらすわけではない
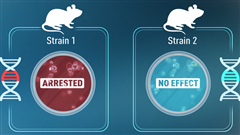
20年前はとてもシンプルに見えました。ゲノムの配列を解析し、病気に関連する変異や変異体のコード配列を調べれば、健康へのロードマップが明確になりました。そこまでシンプルではなかったかもしれませんが、それに近いものでした。 その後の研究により、そのような認識は完全に払拭され、新たな発見のたびに幾層もの難題が見つかりました。ヒトのデータセットにおける重要な発見により、私たちは皆、多くの遺伝子の機能喪失を引き起こす遺伝的変異体を保有していることが明らかになりました。健康なヒト集団には、そのような機能不全遺伝子が平均で約100個あります。複数の近交系(つまり遺伝的にほぼ同一の)マウスの同じ遺伝子をノックアウトすると、個体間で非常にばらつきのある結果が生じる可能性があるため、マウスを使った研究は問題をさらに複雑にしています。非常に極端な場合もあり、生まれることさえできないマウスもいれば、生きているだけでなく基本的に正常に見えるマウスもいます。したがって、遺伝的変異の解釈は各個体の状況によるものであり、一貫して病気やその他の影響を引き起こす浸透性突然変異は一般的ではありません。
4. ほとんどのゲノム変異は構造レベルの変異
ヒトのゲノムはパーセンテージベースでは非常に類似していますが、約32億塩基対の配列に沿った数百万箇所の単一塩基レベルではもちろん異なります。私のゲノムのアデニンはあなたのゲノムのグアニンである可能性があり、それが微妙な変化をもたらし、それが私たちを独自のものにするのです。一塩基多型(SNP)として科学者達に知られているこのような違いが、物語の一部にすぎないことを、私達は20 年前にはほとんど知りませんでした。構造変異体(SV)と呼ばれるその他のより大きな変異体も潜在していますが、実際には、分解されて再構築された配列を変更しないため、ショートリードシーケンス法ではほとんど検出できませんでした。SVでは、ゲノム内の配列に欠失、重複、逆位、挿入がみられ、SNPよりも多くの塩基対の変異が生成されます。一回の読み取りで数十万、さらには数百万の塩基をカバーできる高度なロングリードシーケンス手法が現在、さまざまな疾患にも関連しているこれらのSVの検出と特性解析に使用されています。
5. 遺伝子からメッセンジャーRNA、そしてタンパク質に至るまでの道のりは長く曲がりくねっている
もう何年も前、大学と大学院に通っていた時、私は人生のセントラル・ドグマ(分子生物学の基本的概念)を学びました。A、T、C、G ヌクレオチドで構成される遺伝子は、DNAテンプレートから転写されて、プレメッセンジャーRNA (プレmRNA) を生成します。プレmRNAは処理され、イントロンとして知られる配列が編集され、残りのエクソンが結合されて成熟mRNA になります。次にmRNA は、各細胞内のゲノムが入っている核から、リボソームが存在する細胞質まで移動します。mRNAの3ヌクレオチド配列はそれぞれ特定のアミノ酸をコードしており、翻訳と呼ばれるプロセスでリボソームに1つずつ追加され、最終的に、細胞に必要な機能を実行するために膨大な数の他のタンパク質と結合してタンパク質が生成されます。ただし、それほど単純なことではありません。遺伝子がいつ、どのくらい実際に転写されるかを決定する、目まぐるしい制御ネットワークがあります。プレmRNA の処理は均一ではなく、それらの選択的スプライシングにより、同じ遺伝子からアイソフォームとして知られる多くの異なるタンパク質が生成されます。異常なDNA転写からスプライシング欠陥、翻訳ミスに至るまで、プロセスのどの段階でも問題が発生すると、機能不全や病気が引き起こされる可能性があります。
著者:
Mark Wanner
米国ジャクソン研究所Research Communications部門Associate DirectorのMark Wannerは、ジャクソン研究所の研究に関するコミュニケーションを統括しています。 サイエンスとコミュニケーション両方のバックグラウンドを持つMark Wannerは、さまざまな媒体で生物医学と臨床科学の問題を取り上げ、それらの情報を多くの視聴者層に発信するとともに、その問題について説明しています。
英語原文: What does the future hold for the human genome sequence? (jax.org)